Francesca Bleken from PINK partner SINTEF was authoring in the following publication:
Bleken, F. L.; Jens, K. J.; Solli, K.-A.; and Swang, O. (2025): “CO₂ Utilization through Reaction with Alcohols: A Quantum Chemical Study,” Industrial & Engineering Chemistry Research 2025 64 (46), 22084-22091, DOI: 10.1021/acs.iecr.5c02744
The study by Bleken et al. aligns with the goals of the EU PINK project by demonstrating structured data documentation and methodological transparency, in a manner similar to what is pursued in PINK – starting at the first step of data documentation. This reflects PINK’s ambition to develop methodologies that allow for integration of diverse and hetereogenous datasets and models.
In this study quantum chemical investigation into the mechanisms underlying the reaction between carbon dioxide (CO₂) and alcohols, a central process in carbon capture and utilization (CCU), are provided. The authors note that while such reactions are “roughly thermoneutral, a direct reaction is highly activated,” highlighting the need for catalytic facilitation to achieve feasible kinetics. Their work explores, through density functional theory (DFT), how amines and additional CO₂ molecules act as facilitators that reduce activation barriers and enable the formation of organic carbonates, environmentally benign products relevant to green chemistry, battery electrolytes, and polymer synthesis.
The study builds on the observation that organic carbonates are currently synthesized predominantly via phosgene-based routes, which are toxic and energy-intensive. By contrast, the direct carboxylation of alcohols with CO₂ provides a sustainable pathway, though hindered by kinetic limitations. The authors aim to provide “suggestions for the mechanism(s) of such reactions” and to “determine which of the possible mechanisms is more probable” through quantum chemical modeling of alternative reaction schemes, focusing on methanol as the model alcohol and dimethylamine (DMA) and diisopropylamine (DIPA) as representative amine co-catalysts.
All calculations were performed using DFT (B3LYP/aug-cc-pVDZ) within the NWChem package. Solvent effects were modeled through the conductor-like screening model (COSMO), using dielectric parameters of acetonitrile (ε = 37) to approximate the experimental solvent mixture of methanol, sulfolane, and amine. Transition states were identified via Nudged Elastic Band (NEB) methods and confirmed through frequency analysis, ensuring exactly one imaginary mode per saddle point.
The authors examined several mechanistic pathways (labeled A–D), corresponding respectively to: A) zwitterion formation (CO₂ + amine), B) carbamate formation (proton transfer), C) carbonate formation via alcoholysis, and D) dimethyl carbonate synthesis (condensation of methyl carbonate with methanol). Two variants of mechanism C were considered: amine-facilitated (C‡) and CO₂-facilitated (C*).
Mechanisms A and B were found to be exothermic and weakly activated, forming stable intermediates that align with previous kinetic models of carbamate chemistry. The authors report activation barriers of 6–11 kJ mol⁻¹ for zwitterion formation and 7–15 kJ mol⁻¹ for carbamate formation, depending on the amine used. Notably, “the zwitterion formation is only slightly exothermic and the reverse reaction may occur” (−10 to −17 kJ mol⁻¹), consistent with the transient nature of such intermediates. The key mechanistic insight arises from step C, the conversion of the carbamate intermediate into methyl carbonate. Initial searches for a one-step transition state without facilitators “were fruitless,” since such a four-center mechanism is symmetry-forbidden according to Woodward–Hoffmann rules.
Instead, the inclusion of a CO₂ molecule or an ammonium ion as a cocatalyst was found to enable the reaction. The CO₂-facilitated pathway (C*) emerged as energetically superior, with an activation barrier of 47 kJ mol⁻¹ and a reaction energy of −18 kJ mol⁻¹ for DMA, compared to 158 kJ mol⁻¹ for the amine-facilitated mechanism (C‡). As the authors note, “only the reaction path facilitated with CO₂ exhibits reaction barriers in correspondence with reactions proceeding close to room temperature.” The corresponding barrier for DIPA (66 kJ mol⁻¹) still allowed agreement with experimental data, indicating that the presence of dissolved CO₂ directly determines the kinetics of carbonate formation. These findings provide molecular-level evidence that CO₂ serves a dual function — both as a reactant incorporated into the product and as a reaction facilitator that transiently stabilizes proton transfers. The key transition state (Figure 7 in the paper) shows that “the carbamate functions as a proton acceptor, and the methanol oxygen forms a bond to the new CO₂,” leading to the monomethyl carbonate product.
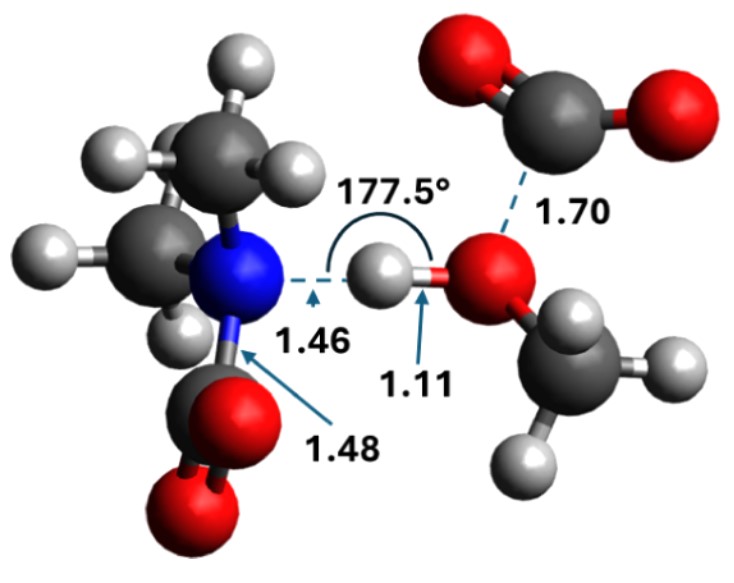
The transition state of the CO2-facilitated mechanism for carbonate formation in dimethylamine. Distances in Angstrom. (Picture (7), taken from publication)
The authors emphasize that the availability of CO₂ in the solvent system is critical. For example, the CO₂ solubility in sulfolane–methanol–amine mixtures was calculated to reach 0.75 mol L⁻¹ at 20 °C, compared to only 0.0001 mol L⁻¹ in pure methanol. Consequently, the reaction feasibility depends not only on intrinsic energetics but also on solvent composition and CO₂ loading, a key insight for designing scalable CCU processes.
Step D, the conversion of methyl carbonate to dimethyl carbonate, proceeds via a proposed SN2 mechanism with an activation barrier of 147 kJ mol⁻¹, consistent with the experimentally observed low yield at room temperature. This high barrier underscores why the first reaction (CO₂ → monomethyl carbonate) is fast and quantitative, while the second condensation step is kinetically limited.
Bleken et al. provide a quantum-chemical validation of a two-CO₂ mechanism for the direct synthesis of organic carbonates from alcohols. The study concludes that “carbon dioxide has a twofold function: (1) as a reactant and (2) as a facilitator for the reaction, by forming a carbamate intermediate that captures the alcoholic proton from the alcohol molecule.” The calculated activation barriers (47–66 kJ mol⁻¹) are consistent with experimental rates observed near room temperature, offering a mechanistic foundation for catalyst and solvent optimization in CCU processes.
Importantly, the authors highlight the industrial and environmental implications of their findings: enabling phosgene-free production of organic carbonates through CO₂ valorisation. The study thus contributes significantly to the rational design of sustainable CO₂ utilization routes, emphasizing that “the solubility of CO₂ should be addressed by proper choice of solvent composition.”
Follow this link to read the full publication.
Parts of the research of this work (SINTEF) has been funded by the European Union`s R&I project PINK (grant agreement # 101137809).